Build Tab
The Build tab is the environment where you can build a molecular structure
How to draw a molecular structure?
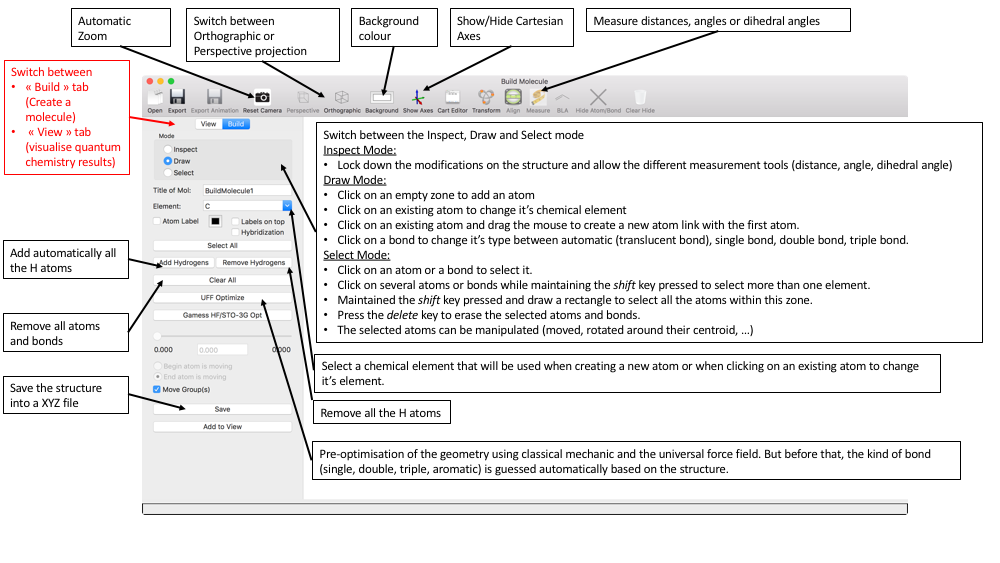
Under the "Build" tab, we have a list of options in order to build a molecular structure:
- Mode "Inspect", "Draw", "Select"
- "Inspect": Locks down the molecular structure and allows to inspect it (mesure distances, angles, dihedral angles, ...)
- "Draw": Allows to add atoms (alone or connected to other atoms by bonds), modify the chemical element of an existing atom, and change the type of bond (single, double or triple bond). This mode is the default one when starting the app.
- "Select": Allows to select one or more atoms (and their bonds) in order to perform some manipulations: delete, move, rotate, duplicate, ...
- Title of Mol: Give a title to your molecule. This title will be used when the structure is saved.
- Element: Change the chemical element. Is used when creating new atoms or when clicking on an existing atom.
- Show atom label checkbox. This label are positioned on the top right corner of each atom by default. They can be located on top of the atom sphere by clicking the Labels on top checkbox. Hybridisation checkbox change the atom label into the hybridisation of the atom as recognised by the Universal Force Field (UFF).
- Clear All: remove all atoms and bonds;
- UFF Optimize: Perform an optimisation of the structure using classical mechanic and the Universal Force Field (UFF).
- Gamess HF/STO-3G Opt: If Gamess US is installed locally and is configure correctly (See the preferences pane of DrawMol app), perform an optimisation of the structure using quantum mechanics and the HF/STO-3G level of theory.
- The slider and text field below allow to tune a bond distance, an angle and a dihedral angles function of the number of atoms selected in the "Select" Mode.
- Save: Save the structure as a xyz file.
- Add to View: Copy the structure in the "View" tab.
Once in the "Draw" mode, you can:
- Add an atom of a given chemical element (chosen from the menu under Element) by clicking on an empty space of the window.
- Add a atom bonded to a previous one by clicking-dragging the mouse from a first atom toward an empty space.
- Add a bond between two existing atoms by clicking-dragging the mouse from a first atom toward the second atom.
- Change the chemical element of an existing atom. To do so, select the appropriate element in the menu Element and click on the atom you want to modify. You can also do a right-click on an existing atom and change the chemical element from the menu that appear (Only a subset of all the atomic elements is available this way).
- Modify the kind of bond (Single, Double, Triple) by clicking on it. At the beginning, the bond is defined as "automatic" (and appear translucent), which means that it's type will be determined when the program add Hydrogen atoms or when it pre-optimise the structure (see below).
- Once the skeleton of the molecule is sketched, the whole structure can be finalise by:
- Clicking on the "Add Hydrogens" button to add all the hydrogen atoms at once.
- Clicking on "UFF Optimize" to do a pre-optimisation using classical mechanic and the universal force field (UFF).
- Both actions trigger an algorithm that automatically determine the hybridisation of the atoms and the kind of bond (Single, Double, Triple, Aromatic) between the atoms. The algorithm bases itself on the molecular structure. For instance, a shorter bond will be assigned to a double or triple bond. Moreover, if a planar 5- or 6-membered ring is sketched, the ring will supposed to be aromatic. We can always force the kind of bond we want by clicking on a translucent bond. The bond will successively becomes single (represented by a single cylinder), double (represented by two cylinders), triple (represented by three cylinders) and back to simple, ...
How to manipulate the molecular structure?
Move the camera
- Rotation: Click-drag the mouse. In the "Draw" mode, be sure to click on an empty space.
- Translation: Drag the mouse while maintaining the shift key pressed. In the "Select" Mode, only the selected atoms are moved
- Zoom: Drag the mouse up or down while maintaining the shift key pressed. The zoom can also be performed from the wheel of the mouse or from the trackpad.
- Roll: Drag the mouse while maintaining the shift-ctrl key pressed. This can also been achieved with two-fingers gesture on the trackpad.
Move some selected atoms and bonds
First, select some atoms using one of the following methods while in the "Select" mode:
- Click on several atoms while maintaining the shift key pressed.
- While maintaining the shift key pressed, drag the mouse to draw a rectangle. All the atoms and bonds within the rectangle are selected.
- Right-click on an atom. In the menu that appear, select either "Add to selection" to add this atom to the selection or "Select All Bonded Atoms" to add this atom and all the atoms that are somehow connected to it to the selection.
- Double-click or force click (with a Force Touch trackpad) on one atom to select all the connected atoms.
The selected atoms (and all the bonds that connect two selected atoms) appear surrounded by a yellow halo. From there, you can:
- Remove these atoms and bonds by pressing the delete key.
- Translate the group of atoms by pressing the ctrl key while dragging the mouse.
- Rotate the group of atoms around their centroid by pressing the alt key while dragging the mouse.
- Duplicate the selected structure by selecting the menu "Molecule" > "Duplicate" (cmd-D shortcut).
- Duplicate the selected structure by applying a precise transformation (translation and rotation) via the "Transform" button on the toolbar.
Right-click on an atom
Save the molecular structure
The molecular structure can be saved as a XYZ file which is nothing else than a text file containing the atomic symbols and the Cartesian coordinates (in Angstrom) for all the atoms. To do so, perform one of the following action:
- Click on the "Save" button.
- Go the "File" > "Save Selected Molecule..." menu. From there, you can select between different file format: H5Mol (the native format of DrawMol), XYZ, CIF, PDB.
A picture of the molecular structure can also be saved via:
- Click on the "Export" button on the toolbar.
- Click on the "File" > "Export" menu (cmd-S shortcut).
Tips
- To undo one or several actions, go to "Edit" > "Undo" menu (cmd-Z shortcut).
- To unselect all the selected atoms, just click on a empty space of the window.
How to create an input file for Gaussian and Gamess-US quantum chemistry packages?
DrawMol has a tool to help you build a input file for various quantum chemistry packages. For now, Gaussian and Gamess-US programs are supported. This tools can be found under "Molecule" > "QM Input Builder" menu.
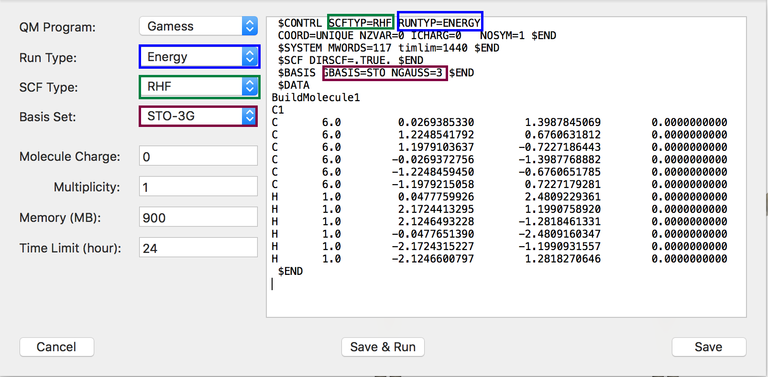
The window is divided into two parts. The right part consist simply in a simple text editor. The left part is giving you several options whose selections are directly mirror on the text shown on the right part. The ideal workflow of this tools is first to select the different options on the left part and then, if needed, to customize the file further by adding/removing manually some text on the right part. The different options on the left parts are:
- The quantum chemistry program for which the input file is created. The choices are Gamess-US and Gaussian.
- The type of calculation such as "Energy", "Optimization", ...
- The method of calculation such as "RHF", "UHF", "MP2" or various exchange-correlation functional of the density functional theory (DFT).
- A given atomic orbital basis set: STO-3G, 6-31G, 6-31G*, ..., cc-pVDZ, aug-cc-pVDZ, ...
- The total charge of the molecule
- The spin multiplicity: 1 (for singlet), 2 (for doublet), 3 (for triplet), ...
- The RAM memory in MB reserved for your job
- If needed, the maximum duration time (in hours) of your job
Execute a Gamess-US job on your local machine
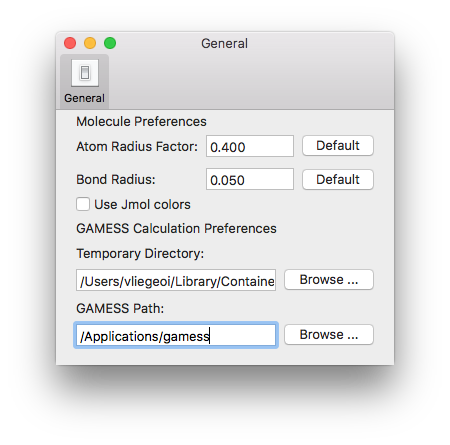
Gamess-US is a freely available quantum chemistry package that can be installed locally on your mac. You just need to register on the Gamess website and then copy the content of the Gamess package into the Applications directory of your mac. To use Gamess program within DrawMol, you first need to check the "DrawMol" > "Preferences" menu and modify the GAMESS Path to be the directory that contains the Gamess executable.
- To execute a calculation on your machine as a background task, click on the "Save & Run" button in the QM input builder tool. Then, chose where the file should be saved on disk and its name and click "Save". The job is directly launch in the background.
- To follow the evolution of your job, go to "Tools" > "QM Status" menu. Once the job is finished, the status becomes "Done".
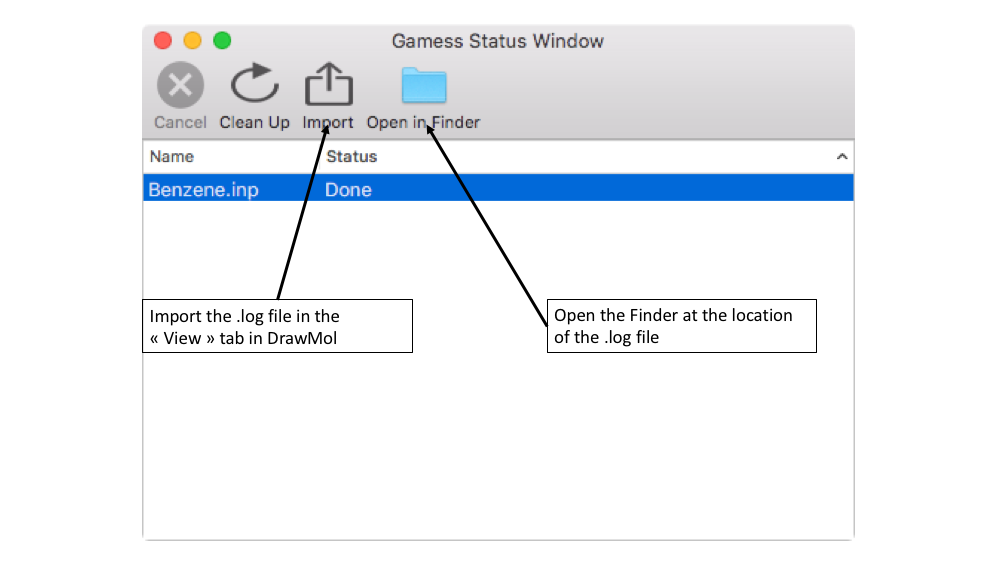
- The final .log file containing all the Gamess results can be found in the same directory where you initially saved the input .inp file.
- TIP 1: From the QM Status window, you can either open the directory where the .log file can be found or directly import the .log file into DrawMol.
- TIP 2: Once the .log file has been imported into DrawMol, you can reused the molecular structure (for instance the optimised structure if the first calculation consists in a geometry optimisation) as the coordinates for a new job by going under "Molecule" > "QM Input Builder" menu
Modify a distance, angle or a dihedral angle
In the "select" mode, when 2, 3 or 4 atoms are selected, you can change the distance, the angle, or the dihedral angle, respectively
You can decide if it's the first or the last selected atom that is going to move
If the "Move Group(s)" option is checked, the groups of atom attached to the moving atom will be displaced as well